
XSB2941 : tumor protein p53 isoform b [Homo sapiens]
[ CaMP Format ]
This entry is computationally expanded from SB0053
* Basic Information
| Organism | Homo sapiens (human) |
| Protein Names | Cellular tumor antigen p53; tumor protein p53 isoform b; p53 tumor suppressor; phosphoprotein p53; p53 antigen; p53 transformation suppressor; transformation-related protein 53 |
| Gene Names | TP53; tumor protein p53 |
| Gene Locus | 17p13.1; chromosome 17 |
| GO Function | Not available |
| Entrez Protein | Entrez Nucleotide | Entrez Gene | UniProt | OMIM | HGNC | HPRD | KEGG |
|---|---|---|---|---|---|---|---|
| NP_001119586 | NM_001126114 | 7157 | Q53GA5_HUMAN | 191170 | 11998 | N/A | hsa:7157 |
* Information From OMIM
Description: The transcription factor p53 responds to diverse cellular stresses to regulate target genes that induce cell cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism. In addition, p53 appears to induce apoptosis through nontranscriptional cytoplasmic processes. In unstressed cells, p53 is kept inactive essentially through the actions of the ubiquitin ligase MDM2 (OMIM:164785), which inhibits p53 transcriptional activity and ubiquitinates p53 to promote its degradation. Numerous posttranslational modifications modulate p53 activity, most notably phosphorylation and acetylation. Several less abundant p53 isoforms also modulate p53 activity. Activity of p53 is ubiquitously lost in human cancer either by mutation of the p53 gene itself or by loss of cell signaling upstream or downstream of p53 (Toledo and Wahl, 2006; Bourdon, 2007; Vousden and Lane, 2007).
Function:
Function: Levine et al. (1991) reviewed p53 function and how alteration or inactivation of p53 by mutation or by interaction with oncogene products of DNA tumor viruses can lead to cancer.
Function: Vogelstein and Kinzler (1992) reviewed function and dysfunction of the p53 gene and outlined 5 mechanisms for p53 inactivation, including disruption of its negative regulator, MDM2 (OMIM:164785).
Function: Science magazine designated p53 the 'Molecule of the Year' for 1993. Culotta and Koshland (1993) and Harris (1993) gave an extensive account of its discovery and elucidation of function, as well as the use of p53 in cancer risk assessment.
Function: Harris and Hollstein (1993) reviewed molecular mechanisms of p53 function and highlighted the clinical implications of changes in the p53 gene in the pathogenesis, diagnosis, prognosis, and therapy of human cancer.
Function: Levine (1997) reviewed all aspects of p53, which he referred to as the cellular gatekeeper for growth and division.
Function: Artandi and Attardi (2005) reviewed the role of p53 in enforcing senescence and apoptotic responses to dysfunctional telomeres. They stated that loss of p53 creates a permissive environment in which critically short telomeres are inappropriately joined to generate chromosomal end-to-end fusions. These fused chromosomes result in cycles of chromosome fusion bridge breakage, which can lead to cancers, especially in epithelial tissues, by facilitating changes in gene copy number.
Function: Toledo and Wahl (2006) reviewed in vitro studies, human tumor data, and mouse models to deduce p53 regulatory mechanisms. They concluded that p53 posttranslational modifications have modulatory roles, whereas MDM2 and MDM4 (OMIM:602704) have more profound roles in p53 regulation.
Function: Bourdon (2007) reviewed p53 isoforms and their roles in p53 regulation and cancer.
Function: Vousden and Lane (2007) reviewed ways in which p53 can contribute to the development, life expectancy, and overall fitness of an organism outside of its role in protecting against cancer development.
Function: Green and Kroemer (2009) reviewed the cytoplasmic functions of p53.
Function:
Function: Fields and Jang (1990), Unger et al. (1992), and Chumakov et al. (1993) discussed the DNA-binding properties of wildtype and mutant p53 and their roles in transcriptional transactivation.
Function: By sequencing 18 human genomic clones that bound p53 in vitro, El-Deiry et al. (1992) identified a consensus binding site with striking internal symmetry, consisting of 2 copies of a 10-bp motif separated by 0 to 13 bp. One copy of the motif was insufficient for p53 binding, and subtle alterations of the motif, even when present in multiple copies, resulted in loss of affinity for p53. Mutants of p53 representing each of the 4 'hotspots' that are altered frequently in human cancers failed to bind the consensus dimer.
Function: Vogelstein and Kinzler (1992) proposed a model in which p53 binds as a tetramer to a p53-binding site (PBS) and activates expression of downstream genes that inhibit growth and/or invasion. Pavletich et al. (1993) stated that tetramerization occurs by interactions between the p53 monomers through a C-terminal domain comprising amino acid residues 325 to 356.
Function: Foster et al. (1999) identified multiple classes of small molecules (300 to 500 daltons) that promoted conformational stability of the wildtype p53 DNA-binding domain and of full-length p53. These compounds also allowed mutant p53 to maintain an active conformation. A prototype compound caused accumulation of conformationally active p53 in cells with mutant p53, enabling it to activate transcription and to slow tumor growth in mice.
Function: Yu et al. (2000) showed that loss of the ERCC6 protein (OMIM:609413) or overexpression of the C-terminal domain of p53 in human cells induced fragility of the RNU1 (OMIM:180680), RNU2 (OMIM:180690), and RN5S (OMIM:180420) genes and the ancient PSU1 locus, which consists entirely of pseudogenes. Moreover, they found that p53 interacted with ERCC6 in vivo and in vitro. The authors proposed that ERCC6 functions as an elongation factor for transcription of structured RNAs, including some mRNAs. Activation of p53 inhibits ERCC6, stalling transcription complexes and locally blocking chromatin condensation.
Function: To determine whether TP53 gene dosage affects transcriptional regulation of target genes, Yoon et al. (2002) performed oligonucleotide array gene expression analysis by using human cells with wildtype p53 or with 1 or both TP53 alleles disrupted by homologous recombination. They identified 35 genes whose expression was significantly correlated with TP53 dosage, including genes involved in signal transduction, cell adhesion, transcription regulation, neurogenesis, and neural crest migration. Motif search analysis revealed that of the genes highly expressed in wildtype and heterozygous p53 cells, several had a putative p53 consensus binding site, suggesting that they may be directly regulated by p53. From these genes, Yoon et al. (2002) chose CSPG2 (OMIM:118661) for further study, and in vitro and in vivo assays showed that CSPG2 was directly transactivated by p53.
Function: Using systems reconstituted with recombinant chromatin templates and coactivators, An et al. (2004) showed that p300 (EP300; OMIM:602700), PRMT1 (OMIM:602950), and CARM1 (OMIM:603934) acted both independently and cooperatively in mediating gene activation by p53. Overexpression of p53 or ultraviolet (UV) irradiation-induced DNA damage in human cell lines led to targeted recruitment of these and other coactivators, as well as accumulation of histone acetylation and methylation marks, on the p53 target gene GADD45 (OMIM:126335).
Function: Bourdon et al. (2005) showed that human p53 and the p53-beta isoform bound differentially to p53-responsive promoters and differentially activated p53-responsive reporter genes. The del133p53 isoform impaired p53-mediated apoptosis. Bourdon et al. (2005) concluded that the functions of p53 are mediated by the interplay between p53 isoforms and full-length p53.
Function:
Function: Using transgenic mice, Lee and Bernstein (1993) found that expression of either of 2 mutant p53 alleles significantly increased cellular resistance of a variety of hematopoietic cell lineages to gamma radiation. They speculated that wildtype p53 may serve as a 'guardian of the genome,' preventing proliferation of a cell that has sustained genetic damage. Thus, cells lacking wildtype p53 protein due to a dominant-negative action of mutant p53 might not undergo radiation-induced cell death, thereby increasing radiation resistance.
Function: El-Deiry et al. (1993) found that induction of WAF1 (CDKN1A; OMIM:116899) was associated with wildtype but not mutant p53 gene expression in a human brain tumor cell line. WAF1 is also called CIP1 or p21, and Harper et al. (1993) showed that it binds to cyclin complexes and inhibits the function of cyclin-dependent kinases. El-Deiry et al. (1993) suggested that p53 is not required for normal development, but its expression is stimulated in certain cellular environments, such as DNA damage or cellular stress. In turn, p53 binds to WAF1 regulatory elements and transcriptionally activates its expression. WAF1 subsequently binds to and inhibits cyclin-dependent kinase activity, preventing phosphorylation of critical cyclin-dependent kinase substrates and blocking cell cycle progression. In tumor cells with inactive p53, this pathway would thereby be defective, permitting unregulated growth.
Function: After DNA damage, many cells appear to enter a sustained arrest in the G2 phase of the cell cycle. Bunz et al. (1998) demonstrated that this arrest could be sustained only when p53 was present in the cell and capable of transcriptionally activating p21. After disruption of either p53 or p21, gamma-radiated cells progressed into mitosis and exhibited G2 DNA content only due to failure of cytokinesis. Bunz et al. (1998) concluded that p53 and p21 are essential for maintaining the G2 checkpoint in human cells.
Function: The centrosome plays a vital role in mitotic fidelity, ensuring establishment of bipolar spindles and balanced chromosome segregation. Centrosome duplication occurs only once during the cell cycle and is therefore highly regulated. Fukasawa et al. (1996) showed that in mouse embryonic fibroblasts lacking p53, multiple copies of functionally competent centrosomes were generated during a single cell cycle. In contrast, mouse embryonic fibroblasts from normal mice or mice deficient in the retinoblastoma tumor suppressor gene product (RB1; OMIM:180200) did not display these abnormalities. The abnormally amplified centrosomes profoundly affected mitotic fidelity, resulting in unequal segregation of chromosomes. These observations implicated p53 in the regulation of centrosome duplication and suggested a possible mechanism by which loss of p53 may cause genetic instability.
Function: Raj et al. (2001) reported that adeno-associated virus (AAV) selectively induced apoptosis in human cells lacking active p53. Cells with intact p53 activity were not killed, but underwent arrest in the G2 phase of the cell cycle. This arrest was characterized by increased p53 activity and p21 levels and by targeted destruction of CDC25C (OMIM:157680). Neither cell killing nor arrest depended upon AAV-encoded proteins. Rather, AAV DNA, which is single stranded with hairpin structures at both ends, elicited in cells a DNA damage response that, in the absence of p53, led to cell death. AAV also inhibited tumor growth in mice. Raj et al. (2001) concluded that viruses can be used to deliver DNA of unusual structure into cells to trigger a DNA damage response without damaging cellular DNA and to selectively eliminate cells lacking p53 activity.
Function: Aylon et al. (2006) found that LATS2 (OMIM:604861) had a role in the p53-dependent G1/S arrest following damage to the mitotic spindle and centrosome dysfunction. LATS2 interacted physically with MDM2 (OMIM:164785) to inhibit p53 ubiquitination and to promote p53 activation.
Function: Xue et al. (2007) used RNA interference to conditionally regulate endogenous p53 expression in a mosaic mouse model of liver carcinoma. Brief reactivation of endogenous p53 in p53-deficient tumors could produce complete tumor regressions. The primary response to p53 was not apoptosis, but instead involved induction of a cellular senescence program associated with differentiation and upregulation of inflammatory cytokines. This program, although producing only cell cycle arrest in vitro, also triggered an innate immune response that targeted tumor cells in vivo, thereby contributing to tumor clearance. Xue et al. (2007) concluded that p53 loss may be required for maintenance of aggressive carcinomas and that the cellular senescence program can act together with the innate immune system to potently limit tumor growth.
Function: Using semiquantitative RT-PCR of wildtype and p53-null mouse embryonic fibroblasts, He et al. (2007) found that expression of miR34a (OMIM:611172), miR34b (OMIM:611374), and miR34c (OMIM:611375) correlated precisely with p53 status. These miR34 genes were direct transcriptional targets of p53 in human and mouse cells, and their induction by DNA damage and oncogenic stress depended on p53 in vitro and in vivo. Ectopic expression of miR34 induced cell cycle arrest in both primary and tumour-derived cell lines, consistent with the ability of miR34 to downregulate a program of genes promoting cell cycle progression.
Function: Qian et al. (2008) identified DEC1 (BHLHB2; OMIM:604256) as a p53 family target gene that mediates p53-induced cellular senescence in response to DNA damage.
Function:
Function: Caelles et al. (1994) developed immortalized somatotropic progenitor cells expressing a temperature-sensitive p53 mutant. In these cells, induction of apoptosis by DNA damage depended strictly on p53 function. Temperature-shift experiments showed that the extent of apoptotic DNA cleavage was directly proportional to the period during which p53 was functional. A shift to the permissive temperature triggered apoptosis following UV radiation-induced DNA damage independently of new RNA or protein synthesis. Caelles et al. (1994) suggested that, rather than activating apoptosis-mediator genes, p53 either represses genes necessary for cell survival or is a component of the enzymatic machinery for apoptotic cleavage or repair of DNA.
Function: Polyak et al. (1997) used serial analysis of gene expression (SAGE) to examine transcripts induced by p53 before the onset of apoptosis. Of 7,202 transcripts identified, only 14 (0.19%) were markedly increased in p53-expressing cells compared with controls. Many of these genes were predicted to encode proteins that could generate or respond to oxidative stress. Additional biochemical and pharmacologic experiments suggested that p53 triggers apoptosis through transcriptional induction of redox-related genes, followed by formation of reactive oxygen species and oxidative degradation of mitochondrial components.
Function: Sablina et al. (2005) found that p53 had an antioxidant function associated with highly responsive p53 target genes induced during nonlethal oxidative stress in several human cell lines. Prooxidant effects of p53 in gravely damaged cells were associated with delayed induction of proapoptotic genes. The p53-dependent increase in reactive oxygen species was secondary to induction of apoptosis and originated from mitochondrial leakage.
Function: Conseiller et al. (1998) constructed a 'chimeric tumor suppressor-1' (CTS1) gene from wildtype p53 by removing the domains that mediate p53 inactivation. CTS1 enhanced transcriptional activity, was resistant to inactivation by MDM2 (OMIM:164785), had the ability to suppress cell growth, and showed faster induction of apoptosis. Conseiller et al. (1998) considered CTS1 to be an alternative for use in gene therapy for wildtype p53-resistant tumors.
Function: Ryan et al. (2000) examined the effect of p53 induction on activation of NF-kappa-B (NFKB; see OMIM:164011), a transcription factor that can protect from or contribute to apoptosis. In human cells without NFKB activity, p53-induced apoptosis was abrogated. Ryan et al. (2000) found that p53 activated NFKB through the RAF (OMIM:164760)/MEK1 (OMIM:176872)/p90(rsk) (see OMIM:601684) pathway rather than the TNFR1 (OMIM:191190)/TRAF2 (OMIM:601895)/IKK (e.g., OMIM:600664) pathway used by TNFA (OMIM:191160). Inhibition of MEK1 blocked p53-induced NFKB activation and apoptosis, but not cell cycle arrest.
Function: Ollmann et al. (2000) identified a Drosophila homolog of p53, which they called Dmp53. Like mammalian p53, Dmp53 bound specifically to human p53-binding sites, and overexpression of Dmp53 induced apoptosis. Inhibition of Dmp53 function rendered cells resistant to x-ray-induced apoptosis, suggesting that Dmp53 is required for the apoptotic response to DNA damage. Unlike mammalian p53, Dmp53 appeared unable to induce a G1 cell cycle block when overexpressed, and inhibition of Dmp53 activity did not affect x-ray-induced cell cycle arrest. These data revealed an ancestral proapoptotic function for p53 and identified Drosophila as an ideal model system for elucidating the p53 apoptotic pathway(s) induced by DNA damage.
Function: Brodsky et al. (2000) also identified a Drosophila p53 homolog and demonstrated that it could activate transcription from a promoter containing binding sites for human p53. Dominant-negative forms of Dmp53 inhibited transactivation in cultured cells and radiation-induced apoptosis in developing tissues. The cis-regulatory region of the proapoptotic gene 'reaper' contains a radiation-inducible enhancer that includes a consensus p53-binding site. Dmp53 could activate transcription from this site in yeast, and a multimer of this site was sufficient for radiation induction in vivo. These results indicated that reaper is a direct transcriptional target of Dmp53 following DNA damage.
Function: Robles et al. (2001) identified a classic p53-responsive element upstream of the APAF1 (OMIM:602233) transcription start site that bound p53 and induced APAF1 gene expression. Apoptosis in a lymphoblastoid cell line, caused by DNA damage due to exposure to ionizing radiation or to doxorubicin, induced APAF1 mRNA and protein expression and was strictly dependent on wildtype p53 function. Robles et al. (2001) concluded that APAF1 is an essential downstream effector of p53-mediated apoptosis.
Function: Fortin et al. (2001) identified 2 p53 consensus binding sites in the mouse Apaf1 promoter and showed that both sites were used by p53. Primary cultures of Apaf1-deficient neurons were significantly protected from p53-induced apoptosis.
Function: Castedo et al. (2001) delineated the apoptotic pathway resulting from human immunodeficiency virus (HIV)-1 envelope glycoprotein (Env)-induced syncytia formation in vitro and in vivo. Immunohistochemical analysis demonstrated the presence of phosphorylated ser15 of p53 as well as the preapoptotic marker tissue transglutaminase (TGM2; OMIM:190196) in syncytium in the apical light zone (T-cell area) of lymph nodes, as well as in peripheral blood mononuclear cells, from HIV-1-positive but not HIV-1-negative donors. The presence of these markers correlated with viral load (HIV-1 RNA levels). Quantitative immunoblot analysis showed that phosphorylation of ser15 of p53 in response to HIV-1 Env was mediated by FRAP (OMIM:601231) and was accompanied by downregulation of protein phosphatase-2A (see OMIM:176915). The phosphorylation was significantly inhibited by rapamycin. Immunofluorescence microscopy indicated that FRAP was enriched in syncytial nuclei and that the nuclear accumulation preceded phosphorylation of ser15 of p53. Castedo et al. (2001) concluded that HIV-1 Env-induced syncytium formation leads to apoptosis via a pathway that involves phosphorylation of ser15 of p53 by FRAP, followed by activation of BAX (OMIM:600040), mitochondrial membrane permeabilization, release of cytochrome C, and caspase activation.
Function: Derry et al. (2001) identified Cep1, a C. elegans homolog of mammalian p53. Cep1 was ubiquitously expressed in embryos, promoted DNA damage-induced apoptosis, and was required for normal meiotic chromosome segregation in the germline. Although somatic apoptosis was unaffected, Cep1 mutants showed hypersensitivity to hypoxia-induced lethality and decreased longevity in response to starvation-induced stress. Overexpression of Cep1 promoted widespread caspase-independent cell death, demonstrating the critical importance of regulating p53 function at appropriate levels.
Function: Sax et al. (2002) presented evidence that BID (OMIM:601997) belongs to a subset of p53-upregulated targets whose induction and subsequent processing mediates p53-induced apoptosis.
Function: Brantley et al. (2002) examined expression of p53 and Rb (OMIM:180200) in 12 eyes containing posterior uveal melanomas following plaque radiotherapy. All cases showed tumor cell loss with residual tumor cells. Strong p53 staining was observed in 6 cases (50%) and was significantly associated with recent radiotherapy. Abnormal cytoplasmic Rb staining was observed in 4 cases (33%). Brantley et al. (2002) concluded that plaque radiotherapy damaged DNA, inhibited cell division, and promoted cell death, at least in part, due to induction of p53.
Function: Seoane et al. (2002) identified MYC (OMIM:190080) as a principal determinant of whether DNA damage-induced activation of p53 results in cell cycle arrest or apoptosis. MYC was directly recruited to the p21 (CDKN1A; OMIM:116899) promoter by the DNA-binding protein MIZ1 (OMIM:604084). This interaction blocked p21 induction by p53 and other activators. As a result, MYC switched the p53-dependent response of colon cancer cells to DNA damage from cytostatic to apoptotic. MYC did not modify the ability of p53 to bind the p21 or PUMA (OMIM:605854) promoters, but it selectively inhibited bound p53 from activating p21 transcription. By inhibiting p21 expression, MYC favored initiation of apoptosis, thereby influencing the outcome of a p53 response in favor of cell death.
Function: Yin et al. (2003) showed that p53 activated transcription of PAC1 (OMIM:603068) by binding to a palindromic site in the PAC1 promoter during apoptosis. PAC1 transcription was induced in response to serum deprivation and oxidative stress, which resulted in p53-dependent apoptosis, but not in response to gamma irradiation, which caused cell cycle arrest. Reduction of PAC1 transcription using small interfering RNA inhibited p53-mediated apoptosis, whereas overexpression of PAC1 increased susceptibility to apoptosis and suppressed tumor formation. Moreover, Yin et al. (2003) found that activation of p53 significantly inhibited MAP kinase (see OMIM:602425) activity. They concluded that, under specific stress conditions, p53 regulates transcription of PAC1 through a novel p53-binding site, and that PAC1 is necessary and sufficient for p53-mediated apoptosis.
Function: Takaoka et al. (2003) found that IFNA (OMIM:147660)/IFNB (OMIM:147640) induced transcription and translation of p53. IFNA/B signaling itself did not activate p53, but it contributed to boosting p53 responses to stress signals. Takaoka et al. (2003) provided examples in which p53 gene induction by IFNA/B contributed to tumor suppression. Furthermore, they showed that p53 was activated in virally infected cells to evoke an apoptotic response and that p53 was critical for antiviral defense of the host. IFNA/B transcriptionally induced p53 through ISGF3 (OMIM:147574). Whereas IFNA/B induced p53 mRNA and increased its protein level, p53-mediated responses such as cell cycle arrest or apoptosis were not observed in cells treated with IFNA/B alone.
Function: Mihara et al. (2003) provided evidence that p53 translocation to mitochondria occurred in vivo in irradiated thymocytes. They showed that p53 could directly induce permeabilization of the outer mitochondrial membrane by forming complexes with the protective BCLXL (see OMIM:600039) and BCL2 (OMIM:151430) proteins, resulting in cytochrome c release. p53 bound BCLXL via its DNA-binding domain. Tumor-derived transactivation-deficient mutants of p53 concomitantly lost the ability to interact with BCLXL and promote cytochrome c release. Mihara et al. (2003) concluded that p53 mutations might represent 'double hits' by abrogating the transcriptional and mitochondrial apoptotic activities of p53.
Function: Chipuk et al. (2004) found that cytosolic localization of endogenous wildtype or transactivation-deficient p53 was necessary and sufficient for apoptosis. p53 directly activated the proapoptotic BCL2 protein BAX in the absence of other proteins to permeabilize mitochondria and engage the apoptotic program. p53 also released both proapoptotic multidomain proteins and BH3-only proteins that were sequestered by BCLXL. Transcription-independent activation of BAX by p53 occurred with similar kinetics and concentrations to those produced by activated BID. Chipuk et al. (2004) proposed that when p53 accumulates in the cytosol, it can function analogously to the BH3-only subset of proapoptotic BCL2 proteins to activate BAX and trigger apoptosis.
Function: Leu et al. (2004) found that after cell stress, p53 interacted with BAK (OMIM:600516), resulting in oligomerization of BAK and release of cytochrome c from mitochondria. Formation of the p53-BAK complex coincided with loss of interaction between BAK and the antiapoptotic protein MCL1 (OMIM:159552). Leu et al. (2004) suggested that p53 and MCL1 have opposing effects on mitochondrial apoptosis by modulating BAK activity.
Function: Liver is generally refractory to apoptosis induced by p53. Leu and George (2007) found that p53 activation led to enhanced expression of IGFBP1 (OMIM:146730) in human hepatoma cells. A portion of intracellular IGFBP1 localized to mitochondria, where it bound the proapoptotic protein BAK. Binding of IGFBP1 to BAK impaired formation of the proapoptotic p53/BAK complex and induction of apoptosis in cultured human and mouse cells and in mouse liver. In contrast, livers of Igfbp1-deficient mice exhibited spontaneous apoptosis accompanied by p53 mitochondrial accumulation and evidence of Bak oligomerization. Leu and George (2007) concluded that IGFBP1 is a negative regulator of the p53/BAK-dependent pathway of apoptosis.
Function: Schultz et al. (2004) showed that TIAF1 (OMIM:609517) and p53 induced apoptosis in human U937 myocytoma cells in both synergistic and antagonistic manners. At optimal levels, both TIAF1 and p53 mediated apoptosis cooperatively. Both proteins also suppressed adherence-independent growth in a mouse fibroblast cell line. In contrast, initiation of apoptosis by overexpressed TIAF1 was blocked by low doses of p53, and vice versa. Ectopic p53 blocked apoptosis in U937 cells stably expressing TIAF1. TIAF1 and p53 did not appear to physically interact; however, nuclear translocation of phosphorylated p53 was significantly reduced in TIAF1-silenced cells. Schultz et al. (2004) concluded that TIAF1 likely participates in the nuclear translocation of activated p53.
Function: Johnson et al. (2005) generated a Trp53 knockin mouse strain carrying mutations of 2 residues crucial for transactivation: leu25 to gln (L25Q) and trp26 to ser (W26S). The mutant protein was designated p53(QS). These mutations had selective effects on the biologic functions of p53 in mouse embryonic fibroblasts. Although its ability to activate various p53 target genes was largely compromised, the p53(QS) protein retained the ability to transactivate Bax. The ability of the p53(QS) protein to elicit a DNA damage-induced G1 cell cycle arrest response was also partially impaired. The p53(QS) protein had selective defects in its ability to induce apoptosis: it was completely unable to activate apoptosis in response to DNA damage and was partially unable to do so when subjected to serum deprivation, but it retained substantial apoptotic activity upon exposure to hypoxia. These findings suggested that p53 acts through distinct, stimulus-specific pathways to induce apoptosis.
Function: Nuclear p53 regulates proapoptotic genes, whereas cytoplasmic p53 directly activates proapoptotic BCL2 proteins to permeabilize mitochondria and initiate apoptosis. Chipuk et al. (2005) found that a tripartite nexus between BCLXL, cytoplasmic p53, and PUMA coordinated these distinct p53 functions in mouse and human cells. After genotoxic stress, BCLXL sequestered cytoplasmic p53. Nuclear p53 caused expression of PUMA, which then displaced p53 from BCLXL, allowing p53 to induce mitochondrial permeabilization. Mutant BCLXL that bound p53, but not PUMA, rendered cells resistant to p53-induced apoptosis irrespective of PUMA expression. Thus, Chipuk et al. (2005) concluded that PUMA couples the nuclear and cytoplasmic proapoptotic functions of p53.
Function: Esteve et al. (2005) found that DNMT1 (OMIM:126375) bound p53 and that the 2 proteins colocalized in nuclei of human colon carcinoma cell lines. DNMT1 and p53 cooperated in methylation and repression of endogenous survivin (BIRC5; OMIM:603352), an antiapoptotic gene containing p53-binding sites in its promoter region.
Function: Raver-Shapira et al. (2007) showed that p53 overexpression in a human colon cancer cell line led to a 20-fold increase in MIR34A (OMIM:611172), paralleling induction of p21. Exposure to whole-body irradiation induced both Mir34a and p21 mRNA in wildtype mice, but it only induced p21 mRNA in p53-knockout mice. Inactivation of MIR34A attenuated p53-mediated apoptosis in cells exposed to genotoxic stress, whereas overexpression of MIR34A mildly increased apoptosis. Independently, Chang et al. (2007) also identified MIR34A as a direct target of p53.
Function: Godar et al. (2008) found that p53 negatively regulated CD44 (OMIM:107269) expression in normal human mammary epithelial cells by binding to a noncanonical p53-binding sequence in the CD44 promoter. Inhibition of CD44 enabled the cells to respond to stress-induced, p53-dependent cytostatic and apoptotic signals that would have otherwise been blocked by CD44. In the absence of p53, CD44 promoted growth in a highly tumorigenic mammary epithelial cell line.
Function:
Function: As normal cells progress toward malignancy, they must switch to an angiogenic phenotype to attract the vasculature that they depend on for growth. Dameron et al. (1994) found that the angiogenic switch in cultured fibroblasts from patients with Li-Fraumeni syndrome (OMIM:151623) coincided with loss of the wildtype p53 allele and resulted from reduced expression of thrombospondin-1 (TSP1, or THBS1; OMIM:188060), a potent inhibitor of angiogenesis. Transfection assays revealed the p53 could stimulate the endogenous TSP1 gene and positively regulate TSP1 promoter sequences. Dameron et al. (1994) concluded that wildtype p53 inhibits angiogenesis in fibroblasts through regulation of TSP1 synthesis.
Function: The earliest genetic alteration in human astrocytoma (see OMIM:137800) progression is mutation of the p53 gene, and one of the earliest phenotypic changes is stimulation of neovascularization. Van Meir et al. (1994) tested the role of p53 in angiogenesis by introducing an inducible wildtype p53 gene into p53-null human glioblastoma cells. The parental cells exhibited strong angiogenic activity, but upon induction of wildtype p53 expression, the cells secreted a factor that could neutralize the angiogenic factors produced by parental cells, as well as the angiogenic activity of FGF2 (OMIM:134920).
Function: Teodoro et al. (2006) showed that p53 transcriptionally activated the alpha-2 collagen prolyl-4-hydroxylase (P4HA1; OMIM:176710) gene, resulting in extracellular release of antiangiogenic fragments of collagen types IV (see OMIM:120130) and XVIII (see OMIM:120328). Conditioned media from cells ectopically expressing either p53 or P4HA1 selectively inhibited growth of primary human endothelial cells. When expressed intracellularly or exogenously delivered, P4HA1 significantly inhibited tumor growth in mice. Teodoro et al. (2006) concluded that there is genetic and biochemical linkage between the p53 tumor suppressor pathway and synthesis of antiangiogenic collagen fragments.
Function: Sano et al. (2007) found that cardiac angiogenesis was crucially involved in the adaptive mechanism of cardiac hypertrophy and that p53 accumulation was essential for transition from cardiac hypertrophy to heart failure. Pressure overload in mice initially promoted vascular growth in the heart by hypoxia-inducible factor-1 (HIF1; see OMIM:603348)-dependent induction of angiogenic factors, and inhibition of angiogenesis prevented development of cardiac hypertrophy and induced systolic dysfunction. Sustained pressure overload induced an accumulation of p53 that inhibited Hif1 activity and thereby impaired cardiac angiogenesis and systolic function. Conversely, promoting cardiac angiogenesis by introducing angiogenic factors or by inhibiting p53 accumulation developed hypertrophy further and restored cardiac dysfunction under chronic pressure overload. Sano et al. (2007) concluded that the antiangiogenic property of p53 may have a crucial function in the transition from cardiac hypertrophy to heart failure.
Function:
Function: Matheu et al. (2007) showed that genetically manipulated mice with increased but otherwise normally regulated levels of Arf (OMIM:600160) and p53 had strong cancer resistance and decreased levels of aging-associated damage. They proposed that the spectra of genes activated by p53 under normal physiologic conditions have a global antioxidant effect, thus decreasing aging-associated oxidative damage.
Function:
Function: Fuchs et al. (1998) stated that direct association of p53 with the cellular protein MDM2 (OMIM:164785) results in ubiquitination and subsequent degradation of p53.
Function: Yin et al. (2002) found that MDM2 induced translation of p53 mRNA from 2 alternative initiation sites, resulting in full-length p53 and an N-terminally truncated protein, p53/47. p53/47 lacks the MDM2-binding site and the most N-terminal transcriptional activation domain of full-length p53. Translation induction required MDM2 to interact directly with the nascent p53 polypeptide and led to a change in the ratio of p53 to p53/47 by inducing translation of both proteins followed by selective degradation of full-length p53.
Function: By mass spectrometry of affinity-purified p53-associated factors, Li et al. (2002) identified the herpesvirus-associated ubiquitin-specific protease (HAUSP; OMIM:602519) as a novel p53-interacting protein. HAUSP strongly stabilized p53, even in the presence of excess MDM2, and induced p53-dependent cell growth repression and apoptosis. HAUSP had an intrinsic enzymatic activity that specifically deubiquitinated p53 both in vivo and in vitro. Expression of a catalytically inactive point mutation of HAUSP in cells increased the levels of p53 ubiquitination and also destabilized p53. Li et al. (2002) concluded that p53 can be stabilized by direct deubiquitination and suggested that HAUSP may function as a tumor suppressor in vivo through stabilization of p53.
Function: Both p53 and MDM2 interact with p300 (EP300; OMIM:602700)/CREB-binding protein (CBP; OMIM:600140) transcriptional coactivators. Grossman et al. (2003) observed that purified p300 exhibited intrinsic ubiquitin ligase activity. In vitro, p300 with MDM2 catalyzed p53 polyubiquitination, whereas MDM2 alone catalyzed p53 monoubiquitination. Grossman et al. (2003) concluded that generation of the polyubiquitinated forms of p53 that are targeted for proteasome degradation requires the intrinsic ubiquitin ligase activities of MDM2 and p300.
Function: Colaluca et al. (2008) described a previously unknown function for human NUMB (OMIM:603728) as a regulator of tumor protein p53. NUMB enters in a tricomplex with p53 and the E3 ubiquitin ligase MDM2 thereby preventing ubiquitination and degradation of p53. This results in increased p53 protein levels and activity, and in regulation of p53-dependent phenotypes. In breast cancers there is frequent loss of NUMB expression. Colaluca et al. (2008) showed that, in primary breast tumor cells, this event causes decreased p53 levels and increased chemoresistance. In breast cancers, loss of NUMB expression causes increased activity of the receptor Notch (OMIM:190198). Thus, in these cancers, a single event--loss of NUMB expression--determines activation of an oncogene (NOTCH1) and attenuation of the p53 tumor suppressor pathway. Biologically, this results in an aggressive tumor phenotype, as witnessed by findings that NUMB-defective breast tumors display poor prognosis.
Function: Le Cam et al. (2006) found that human E4F1 (OMIM:603022) functioned as as a ubiquitin E3 ligase for p53 both in vitro and in vivo. E4F1-mediated ubiquitylation of p53 occurred at sites distinct from those targeted by MDM2, competed with PCAF (OMIM:602303)-induced acetylation of p53, and did not target p53 for proteasomal degradation. E4F1-stimulated p53-ubiquitin conjugates were associated with chromatin, and their stimulation coincided with induction of a p53-dependent transcriptional program specifically involved in cell cycle arrest, but not apoptosis. Le Cam et al. (2006) concluded that E4F1 is a key posttranslational regulator of p53 that plays an important role in the cellular life-or-death decision controlled by p53.
Function:
Function: Shieh et al. (1997) showed that, upon DNA damage, p53 was phosphorylated at ser15 and that this event led to reduced interaction of p53 with its negative regulator, MDM2 (OMIM:164785). Furthermore, phosphorylation of p53 at ser15 and ser37 by purified DNA-dependent protein kinase (see OMIM:600899) impaired the ability of MDM2 to inhibit p53-dependent transactivation. Shieh et al. (1997) concluded that these effects were most likely due to a conformational change induced by phosphorylation of p53. Shieh et al. (1997) proposed that under normal unstressed conditions, p53 associates with MDM2, and p53-dependent transactivation is repressed. Upon DNA damage, p53 is phosphorylated at ser15, which induces a conformational change that makes MDM2 unable to bind p53, relieving the inhibitory effect of MDM2 on p53.
Function: Oda et al. (2000) identified an apoptosis-inducing gene, p53AIP1 (OMIM:605426), whose expression was induced by wildtype p53. Upon severe DNA damage, ser46 on p53 was phosphorylated, leading to induction of apoptosis. Substitution of ser46 inhibited the ability of p53 to induce apoptosis and selectively blocked expression of p53AIP1. Oda et al. (2000) concluded that p53AIP1 mediates p53-dependent apoptosis and that phosphorylation of ser46 on p53 regulates transcriptional activation of p53AIP1.
Function: Okamura et al. (2001) found that overexpression of P53DINP1 (OMIM:606185) and DNA damage induced by double-strand breaks synergistically enhanced ser46 phosphorylation of p53, induction of p53AIP1, and apoptotic cell death. P53DINP1 interacted with a protein complex that phosphorylated p53 on ser46.
Function: Hirao et al. (2000) found that Chk2 (OMIM:604373) -/- mouse embryonic cells were defective for p53 stabilization and for induction of p53-dependent transcripts, such as p21, in response to gamma irradiation. Reintroduction of the Chk2 gene restored p53-dependent transcription in response to gamma irradiation. Human CHK2 directly phosphorylated p53 on ser20, a modification known to interfere with MDM2 binding. Hirao et al. (2000) concluded that phosphorylation of p53 by CHK2 increases p53 stability by preventing ubiquitination in response to DNA damage. The results provided a mechanistic link between CHK2 and p53 to explain the phenotypic similarity of Li-Fraumeni syndrome-1 (LFS1; OMIM:151623), which is caused by mutations in p53, and Li-Fraumeni syndrome-2 (LFS2; OMIM:609265), which is caused by mutations in CHK2.
Function: Phosphorylation of the human p53 protein at ser392 is responsive to UV but not gamma irradiation. Keller et al. (2001) identified and purified a mammalian UV-activated protein kinase complex that phosphorylated ser392 in vitro. This kinase complex contained casein kinase-2 (CK2; see OMIM:115441) and the chromatin transcriptional elongation factor FACT, a heterodimer of SPT16 (OMIM:605012) and SSRP1 (OMIM:604328). In vitro studies showed that FACT altered the specificity of CK2 in the complex such that it selectively phosphorylated p53 over other substrates, including casein, and phosphorylation by the kinase complex enhanced p53 activity.
Function: Zhang and Xiong (2001) identified a nuclear export signal in the N terminus of p53 containing 2 serines that were phosphorylated after DNA damage. The N-terminal signal was required for p53 nuclear export in collaboration with the C-terminal nuclear export signal. Serine-15-phosphorylated p53 induced by UV irradiation was not exported. Zhang and Xiong (2001) concluded that DNA damage-induced phosphorylation may achieve optimal p53 activation by inhibiting both MDM2 binding to, and nuclear export of, p53.
Function: Expression of oncogenic RAS (HRAS; OMIM:190020) mutants, such as HRASV12 (see OMIM:190020.0001), in primary human cells activates p53, thereby protecting cells from transformation. Bulavin et al. (2002) showed that p38 MAPK (MAPK14; OMIM:600289) phosphorylated p53 at ser33 and ser46 in a human fibroblast cell line expressing oncogenic RAS. The activity of p38 MAPK was regulated by the p53-inducible phosphatase PPM1D (OMIM:605100), creating a potential feedback loop. Expression of oncogenic Ras suppressed PPM1D mRNA induction, leaving p53 phosphorylated at ser33 and ser46 and in an active state. Overexpression of PPM1D reduced p53 phosphorylation at these sites, which abrogated RAS-induced apoptosis and partially rescued cells from cell-cycle arrest.
Function: Hofmann et al. (2002) and D'Orazi et al. (2002) found that HIPK2 (OMIM:606868) colocalized and interacted with p53 and CBP (CREBBP; OMIM:600140) within promyelocytic leukemia nuclear bodies. Activation of HIPK2 by UV radiation led to phosphorylation of p53 at ser46, facilitating CBP-mediated acetylation of p53 at lys382 and promoting p53-dependent gene expression.
Function: Rinaldo et al. (2007) stated that phosphorylation of p53 on ser46 shifts the affinity of p53 for promoters of genes involved in cell cycle arrest to promoters of genes involved in apoptosis. They observed that lethal DNA damage increased HIPK2 expression, whereas sublethal DNA damage repressed HIPK2 expression. Rinaldo et al. (2007) identified HIPK2 as a target for MDM2-mediated ubiquitin-dependent degradation and found that HIPK2 degradation only occurred in growth-arresting conditions when MDM2 was efficiently induced by p53.
Function: Taira et al. (2007) found that DYRK2 (OMIM:603496) phosphorylated p53 on ser46 in vitro and in human cells. Upon exposure to genotoxic stress, DYRK2 translocated into the nucleus and phosphorylated p53 on ser46, inducing P53AIP1 expression and apoptosis in a ser46 phosphorylation-dependent manner.
Function: Cordenonsi et al. (2007) found that RTK/Ras/MAPK activity induced p53 N-terminal phosphorylation, enabling interaction of p53 with TGF-beta (OMIM:190180)-activated SMADs (see OMIM:601595). This mechanism confined mesoderm specification in Xenopus embryos and promoted TGF-beta cytostasis in human cells.
Function:
Function: Luo et al. (2000) found that deacetylation of p53 was mediated by a histone deacetylase-1 (HDAC1; OMIM:601241)-containing complex, and they purified a p53 target protein, MTA1L1 (OMIM:603947), in the deacetylase complexes. MTA1L1, a component of the nucleosome remodeling and histone deacetylation (NURD) complex, specifically interacted with p53 in vitro and in vivo. Expression of MTA1L1 reduced steady-state levels of acetylated p53, repressed p53-dependent transcriptional activation, and modulated p53-mediated cell growth arrest and apoptosis. Luo et al. (2000) concluded that deacetylation and functional interactions between the MTA1L1-associated NURD complex may represent an important pathway to regulate p53 function.
Function: Pearson et al. (2000) found that the tumor suppressor PML (OMIM:102578) regulated the p53 response to oncogenic signals. Oncogenic RAS (HRAS; OMIM:190020) upregulated PML expression, and overexpression of PML induced senescence in a p53-dependent manner. p53 was acetylated at lys382 upon RAS expression, an event essential for its biologic function. RAS induced relocalization of p53 and the CBP (CREBBP; OMIM:600140) acetyltransferase within PML nuclear bodies and induced formation of a trimeric p53-PML-CBP complex. RAS-induced p53 acetylation, p53-CBP complex stabilization, and senescence were lost in PML -/- fibroblasts. Pearson et al. (2000) concluded that their there is a link between PML and p53 and that integrity of PML bodies is required for p53 acetylation and senescence upon oncogene expression.
Function: Vaziri et al. (2001) found that SIRT1 (OMIM:604479) bound and deacetylated p53 specifically at lys382, modification of which is implicated in activation of p53 as a transcription factor. Expression of wildtype SIRT1 in human cells reduced p53 transcriptional activity. In contrast, expression of a catalytically inactive SIRT1 protein potentiated p53-dependent apoptosis and radiosensitivity.
Function: Luo et al. (2001) found that nicotinamide (vitamin B3) inhibited NAD-dependent p53 deacetylation induced by SIRT1 and also enhanced p53 acetylation levels in vivo. SIRT1 repressed p53-dependent apoptosis in response to DNA damage and oxidative stress, whereas expression of a SIRT1 point mutant increased the sensitivity of cells in the stress response.
Function: Using a yeast p53 dissociator assay with a HeLa cell expression library, Wang et al. (2001) identified ADA3 (TADA3L; OMIM:602945), a part of histone acetyltransferase (HAT) complexes, as a cofactor for p53 activity. ADA3 and p53 interacted directly in cotransfected cells. Mutation analysis showed that the N terminus of ADA3 interacted with the N terminus of p53, while the C terminus of ADA3 interacted with ADA2 (TADA2L; OMIM:602276) and p300 (EP300; OMIM:602700), components of HAT complexes. Following DNA damage, p53 was phosphorylated at its N terminus, and this enhanced the amount of p53 that could be coimmunoprecipitated with ADA3. The N terminus of ADA3 alone could inhibit p53 transcriptional activity and prevent p53-mediated apoptosis. Wang et al. (2001) concluded that ADA3 function is essential for full transcriptional activity of p53 and p53-mediated apoptosis.
Function: Tang et al. (2006) found that lys120 (K120) within the DNA-binding domain of p53 was acetylated in several human cell lines, and that acetylation of K120 was significantly enhanced upon DNA damage. This modification of p53 was catalyzed by TIP60 (OMIM:601409). A tumor-derived p53 mutant defective for TIP60-mediated acetylation, lys120 to arg (K120R), abrogated p53-dependent activation of apoptosis but had no significant effect on cell growth arrest.
Function: Sykes et al. (2006) showed that the p53 K120R mutation selectively blocked transcription of proapoptotic target genes such as BAX (OMIM:600040) and PUMA (OMIM:605854). Depletion of TIP60 or MOF (MYST1; OMIM:609912), another enzyme that can acetylate p53 at K120, inhibited the ability of p53 to activate BAX and PUMA transcription. Sykes et al. (2006) showed that the acetyl-K120 form of p53 specifically accumulated at proapoptotic target genes.
Function: Upon DNA damage, p53 is acetylated by CBP at K373 and K382, by PCAF (OMIM:602303) at K320, and by TIP60 at K120. This acetylation enhances the ability of p53 to bind DNA and recruit transcriptional coactivators to p53-responsive promoters. Li et al. (2007) showed that acetylation of K373 and K382 on p53 led to their direct interaction with the tandem bromodomains of TAF1 (OMIM:313650). p53 recruited TAF1 to a distal p53-binding site on the p21 (CDKN1A; OMIM:116899) promoter prior to the DNA looping that brings TAF1 to the TATA box-containing core promoter.
Function: Tang et al. (2008) identified K164 as an additional site for acetylation of full-length human p53 by CBP/p300. K164 is a conserved residue located in the L2 loop of the DNA-binding core domain of p53. Although acetylation defects at each individual site (K164, K120, and the 6 C-terminal lysines) could be compensated by acetylation at other sites, loss of acetylation at all of these major sites completely abolished the ability of p53 to activate p21 and suppress cell growth. Acetylation blocked the interaction of p53 with its repressors MDM2 and MDMX (MDM4; OMIM:602704) on the p21 promoter, and this directly resulted in p53 activation regardless of its phosphorylation status. In addition, inactivation of MDM2 and MDMX restored the transcriptional functions of unacetylated p53.
Function:
Function: Chuikov et al. (2004) reported that SET9 (OMIM:606594) specifically methylated p53 at lys372 within the C-terminal regulatory region in human cells. Methylated p53 was restricted to the nucleus, and the modification stabilized p53. SET9 regulated expression of p53 target genes in a manner dependent on the p53 methylation site.
Function: Huang et al. (2006) reported that SMYD2 (OMIM:610663) methylated lys370 in p53. In contrast to methylation of lys372, methylation of lys370 repressed p53-mediated transcriptional regulation by maintaining low concentrations of promoter-associated p53. Reduction of SMYD2 by siRNA enhanced p53-mediated apoptosis. SET9-mediated methylation of lys372 inhibited SMYD2-mediated methylation of lys370, in part, by blocking interaction between p53 and SMYD2. Huang et al. (2006) concluded that, similar to histones, p53 is subject to both activating and repressing lysine methylation.
Function: Huang et al. (2007) demonstrated that in human cells the histone lysine-specific demethylase LSD1 (OMIM:609132) interacts with p53 to repress p53-mediated transcriptional activation, and to inhibit the role of p53 in promoting apoptosis. They found that in vitro, LSD1 removes both monomethylation (K370me1) and dimethylation (K370me2) at K370, a SMYD2-dependent monomethylation site (Huang et al., 2006). However, in vivo, LSD1 showed a strong preference to reverse K370me2, which is performed by a distinct methyltransferase. Huang et al. (2007) concluded that K370me2 has a different role in regulating p53 from that of K370me1: K370me1 represses p53 function, whereas K370me2 promotes association with the coactivator 53BP1 (OMIM:605230). The observations of Huang et al. (2007) showed that p53 is dynamically regulated by lysine methylation and demethylation and that the methylation status at a single lysine residue confers distinct regulatory output.
Function: Shi et al. (2007) showed that SET8 (SETD8; OMIM:607240) monomethylated p53 in human cell lines. This monomethylation suppressed p53-mediated transcriptional activation of highly responsive target genes, such as p21 (CDKN1A; OMIM:116899) and PUMA (BBC3; OMIM:605854), but it had little influence on weak p53 targets. Depletion of SET8 augmented the proapoptotic and checkpoint activation functions of p53, and SET8 expression was downregulated upon DNA damage.
Function:
Function: By immunoprecipitation and binding analyses, Lu and Levine (1995) showed that TAF9 (OMIM:600822) interacted with the N-terminal domain of p53 at sites identical to those bound by MDM2 (OMIM:164785). Antibodies to TAF9 inhibited p53-activated transcription. Lu and Levine (1995) concluded that p53 activity is regulated by MDM2 and TAF9 competing for the same region of the p53 protein.
Function: Based on evidence for JNK (OMIM:602896) association with p53, Fuchs et al. (1998) sought to elucidate the role of nonactive JNK2 in regulating p53 stability. The amount of p53-JNK complex was inversely correlated with the p53 level in nonstressed mouse fibroblasts. A peptide corresponding to the JNK-binding site on p53 inhibited JNK binding and efficiently blocked ubiquitination of p53. Similarly, p53 lacking the JNK-binding site exhibited a longer half-life than wildtype p53. Outcompeting JNK association with p53 increased the level of p53, whereas overexpression of a phosphorylation mutant form of JNK inhibited p53 accumulation. JNK-p53 and MDM2-p53 complexes were preferentially found in G0/G1 and S/G2M phases of the cell cycle, respectively. Fuchs et al. (1998) concluded that JNK is an MDM2-independent regulator of p53 stability in nonstressed cells.
Function: Bernal et al. (2002) identified securin (PTTG1; OMIM:604147) as a negative regulator of p53. Assays demonstrated that p53 interacted specifically with securin both in vitro and in vivo, and this interaction blocked specific binding of p53 to DNA and inhibited its transcriptional activity. Securin also inhibited the ability of p53 to induce cell death. Transfection of human non-small cell lung carcinoma cells with securin induced an accumulation of cells in G2 that compensated for the loss of G2 cells caused by transfection with p53. Both apoptotic and transactivating functions of p53 were potentiated in securin-deficient human tumor cells cells compared with parental cells.
Function: Zacchi et al. (2002) found that, on DNA damage, p53 interacted with PIN1 (OMIM:601052), a peptidyl-prolyl isomerase that regulates proteins involved in cell cycle control and apoptosis. The interaction was strictly dependent on DNA damage-induced p53 phosphorylation and required ser33, thr81, and ser315. On binding, PIN1 generated conformational changes in p53 that enhanced its transactivation activity. Stabilization of p53 was impaired in UV-treated Pin1 -/- mouse cells owing to the inability of p53 to efficiently dissociate from MDM2. As a consequence, Pin1 -/- cells exhibited a reduced p53-dependent response to DNA damage that correlated with diminished transcriptional activation of p53-regulated genes. Zheng et al. (2002) presented similar findings and showed that PIN1-mediated p53 activation required the WW domain and isomerase activity of PIN1.
Function: Fernandez-Fernandez et al. (2005) found that S100B (OMIM:176990) and S100A4 (OMIM:114210) bound the C-terminal tetramerization domain of p53 when the domain was exposed in lower oligomerization states, disrupting p53 tetramerization. S100B also bound to the negative regulatory and nuclear localization domains of p53, resulting in tight binding. Because trafficking of p53 depends on its oligomerization state, Fernandez-Fernandez et al. (2005) proposed that S100B and S100A4 may regulate subcellular localization of p53 but with different effects on p53 function in cell cycle control due to their differences in binding p53.
Function: Barral et al. (2005) showed that E1BAP5 (HNRNPUL1; OMIM:605800), a heterogeneous nuclear ribonucleoprotein family member, interacted directly with p53 and inhibited induction of p53-regulated genes following UV irradiation.
Function:
Function: Chen et al. (1990) introduced single copies of exogenous p53 genes containing either point-mutated or wildtype versions of the p53 cDNA sequence into a human osteosarcoma cell line lacking endogenous p53 by infecting the cells with recombinant retroviruses. Expression of wildtype p53 suppressed the neoplastic phenotype. In a 2-allele configuration, wildtype p53 was phenotypically dominant to mutated p53.
Function: Halevy et al. (1990) demonstrated that the ability of a p53 mutant to bind endogenous p53 is not the sole determinant of its oncogenic potential. They concluded that p53 mutants involved in the neoplastic process display various properties, including gain of function.
Function: In tumors showing rapid growth, hexokinase-2 (HK2; OMIM:601125) is highly expressed to facilitate high rates of glucose catabolism, which promote rapid tumor proliferation. Mathupala et al. (1997) cloned p53 from the AS-30D rat hepatoma cell line and identified 2 point mutations at the periphery of the p53 core DNA-binding domain. Using coexpression studies, they showed that overexpressed mutant p53 significantly and reproducibly activated the HK2 promoter and increased gene expression. The findings were consistent with reports describing the transactivating effects of p53 on various genes (Unger et al., 1992; Chumakov et al., 1993; Zhang et al., 1993), but they contrasted with reports that mutant p53 functions in tumor cells only to prevent wildtype p53 from transactivating genes involved in suppressing cell proliferation (Fields and Jang, 1990; Farmer et al., 1992).
Function: Raman et al. (2000) found low p53 mRNA levels in a large proportion of breast tumors. They identified consensus HOX-binding sites in the p53 promoter and found that transient transfection of HOXA5 (OMIM:142952) activated the p53 promoter. Expression of HOXA5 in epithelial cancer cells expressing wildtype p53, but not in isogenic variants lacking p53, led to apoptotic cell death. Moreover, breast cancer cell lines and patient tumors displayed a coordinate loss of p53 and HOXA5 mRNA and protein expression. The HOXA5 promoter region was methylated in 16 of 20 p53-negative breast tumor specimens. Raman et al. (2000) concluded that loss of p53 expression in human breast cancer may be primarily due to lack of HOXA5 expression.
Function: Constitutive activation of JAK2 (OMIM:147796) is frequently detected in human cancers. Reid et al. (2004) found that reintroduction of p53 in 2 human ovarian cancer cell lines with mutant p53 and high levels of phosphorylated JAK2 upregulated protein tyrosine phosphatase-1B (PTPN1; OMIM:176885), reduced JAK2 tyrosine phosphorylation, and induced apoptosis.
Function: Using mouse and human cells, Insinga et al. (2004) showed that the acute promyelocytic leukemia-associated fusion proteins PML/RAR (see PML; OMIM:102578) and PLZF/RAR (see ZNF145; OMIM:176797) directly inhibited p53, allowing leukemic blasts to evade p53-dependent cancer surveillance pathways. PML/RAR expression led to p53 deacetylation and destabilization, resulting in MDM2 (OMIM:164785)-mediated p53 degradation. Protection of PML/RAR-expressing cells from the p53-dependent genotoxic stress response depended on the presence of wildtype PML, suggesting that PML/RAR acts as a gain-of-function mutation.
Function: Bartkova et al. (2005) showed that in clinical specimens from different stages of human tumors of urinary bladder, breast, lung, and colon, the early precursor lesions, but not normal tissues, commonly expressed markers of an activated DNA damage response. These included phosphorylated kinases ATM (OMIM:607585) and CHK2 (OMIM:604373) and phosphorylated histone H2AX (OMIM:601772) and p53. Similar checkpoint responses were induced in cultured cells upon expression of different oncogenes that deregulate DNA replication. Together with genetic analyses, including a genomewide assessment of allelic imbalances, Bartkova et al. (2005) concluded that early in tumorigenesis, before genomic instability and malignant conversion, human cells activate an ATR/ATM-regulated DNA damage response network that delays or prevents cancer. Mutations compromising this checkpoint, including defects in the ATM-CHK2-p53 pathway, might allow cell proliferation, survival, increased genomic instability, and tumor progression.
Function: Gorgoulis et al. (2005) analyzed a panel of human lung hyperplasias that retained wildtype p53 genes and had no signs of gross chromosomal instability and found signs of a DNA damage response, including histone H2AX and CHK2 phosphorylation, p53 accumulation, focal staining of p53 binding protein-1 (53BP1; OMIM:605230), and apoptosis. Progression to carcinoma was associated with p53 or 53BP1 inactivation and decreased apoptosis. A DNA damage response was also observed in dysplastic nevi and in human skin xenografts, in which hyperplasia was induced by overexpression of growth factors. Both lung and experimentally-induced skin hyperplasias showed allelic imbalance at loci prone to DNA double-strand break formation when DNA replication is compromised (common fragile sites). Gorgoulis et al. (2005) proposed that, from its earliest stages, cancer development is associated with DNA replication stress, which leads to DNA double-strand breaks, genomic instability, and selective pressure for p53 mutations.
Function: Fujiwara et al. (2005) transiently blocked cytokinesis in p53-null mouse mammary epithelial cells, enabling isolation of diploid and tetraploid cultures. Tetraploid cells had an increased frequency of whole-chromosome missegregation and chromosomal rearrangements, and only tetraploid cells were transformed in vitro after exposure to carcinogen. In the absence of carcinogen, only tetraploid cells gave rise to malignant mammary epithelial cancers when transplanted subcutaneously into nude mice. These tumors all contained numerous nonreciprocal translocations and an 8- to 30-fold amplification of a chromosomal region containing a cluster of matrix metalloproteinase (MMP) genes, overexpression of which had been linked to mammary tumors in humans and in animal models (Egeblad and Werb, 2002). Fujiwara et al. (2005) concluded that tetraploidy enhances the frequency of chromosomal alterations and promotes tumor development in p53-null mouse mammary epithelial cells.
Function: Chen et al. (2005) showed that conditional inactivation of Trp53 in mouse prostate failed to produce a tumor phenotype, whereas complete Pten (OMIM:601728) inactivation in prostate triggered nonlethal invasive prostate cancer after long latency. Strikingly, combined inactivation of Pten and Trp53 elicited invasive prostate cancer as early as 2 weeks after puberty and was invariably lethal by 7 months of age. Acute Pten inactivation induced growth arrest through the p53-dependent cellular senescence pathway both in vitro and in vivo, which could be fully rescued by combined loss of Trp53. In addition, Chen et al. (2005) detected evidence of cellular senescence in specimens from early-stage human prostate cancer. They concluded that cellular senescence plays a role in restricting tumorigenesis in vivo and that p53 is an essential failsafe protein of Pten-deficient tumors.
Function: Laurie et al. (2006) showed that the tumor surveillance pathway mediated by ARF (see OMIM:600160), MDM2, MDMX (OMIM:602704), and p53 was activated after loss of RB1 during retinogenesis in mouse and human. RB1-deficient retinoblasts underwent p53-mediated apoptosis and exited the cell cycle. Subsequently, amplification of the MDMX gene and increased expression of MDMX protein were strongly selected for during tumor progression as a mechanism to suppress the p53 response in RB1-deficient retinal cells. Laurie et al. (2006) concluded that the p53 pathway is inactivated in retinoblastoma and that this cancer does not originate from intrinsically death-resistant cells, as had been thought.
Function: Matoba et al. (2006) found that p53 modulated the balance between the use of respiratory and glycolytic pathways. They identified SCO2 (OMIM:604272), which is critical for regulating the COX complex, the major site of oxygen use in the eukaryotic cell, as the downstream mediator of this effect in mice and human cancer cell lines. Disruption of the SCO2 gene in human cancer cells with wildtype p53 recapitulated the metabolic switch toward glycolysis exhibited by p53-deficient cells. Matoba et al. (2006) concluded that the coupling of p53 to mitochondrial respiration by SCO2 provides a possible explanation for the Warburg effect, in which cancer cells preferentially use glycolytic pathways for energy generation while downregulating their aerobic respiratory activity.
Function: Ventura et al. (2007) showed that restoring endogenous p53 expression led to regression of autochthonous lymphomas and sarcomas in mice without affecting normal tissues. The main consequence of p53 restoration was apoptosis in lymphomas and suppression of cell growth with features of cellular senescence in sarcomas. Ventura et al. (2007) concluded that sustained p53 inactivation is required for tumor maintenance.
Function: Feng et al. (2007) found evidence that increased tumor incidence with age may be due to reduced p53 function in older populations. They showed that p53 responses to gamma irradiation and other stresses were reduced in aging mice and in cultured splenocytes from older mice, which included decreased p53 transcriptional activity and p53-dependent apoptosis. The function of Atm declined significantly with age, which may be responsible for reduced p53 activity. The time of onset of decreased p53 response correlated with the life span of mice; mice that lived longer delayed their onset of decreased p53 activity.
Function: Foo et al. (2007) noted that only about one-half of cancers have p53 loss-of-function mutations. They demonstrated that the apoptotic function of wildtype p53 was inactivated by binding to ARC (NOL3; OMIM:605235) in the nucleus of human cancer cell lines. ARC bound to the p53 tetramerization domain, which inhibited p53 tetramerization and exposed a nuclear export signal in p53, leading to CRM1 (XPO1; OMIM:602559)-dependent relocation of p53 to the cytoplasm. Knockdown of endogenous ARC in breast cancer cells resulted in spontaneous tetramerization of endogenous p53, accumulation of p53 in the nucleus, and activation of endogenous p53 target genes. In primary human breast cancers with nuclear ARC, p53 was almost always wildtype. Conversely, nearly all breast cancers with mutant p53 lacked nuclear ARC. Foo et al. (2007) concluded that nuclear ARC is induced in cancer cells and negatively regulates p53.
Function: The Cancer Genome Atlas Research Network (2008) reported the interim integrative analysis of DNA copy number, gene expression, and DNA methylation aberrations in 206 glioblastomas (OMIM:137800) and nucleotide sequence alterations in 91 of the 206 glioblastomas. The authors found that p53 itself showed mutation or homozygous deletion in 35% of tumors and that there was altered p53 signaling in 87% of tumors, as demonstrated by homozygous deletion or mutations in CDKN2A in 49% of tumors, amplification of MDM2 in 14%, and amplification of MDM4 in 7%.
Function: Zheng et al. (2008) showed that concomitant central nervous system-specific deletion of p53 and Pten in the mouse central nervous system generates a penetrant acute-onset high grade malignant glioma phenotype with notable clinical, pathologic, and molecular resemblance to primary glioblastoma in humans. This genetic observation prompted TP53 and PTEN mutation analysis in human primary glioblastoma, demonstrating unexpectedly frequent inactivating mutations of TP53 as well as the expected PTEN mutations. Integrated transcriptomic profiling, in silico promoter analysis, and functional studies of murine neural stem cells established that dual, but not singular, inactivation of p53 and Pten promotes an undifferentiated state with high renewal potential and drives increased Myc (OMIM:190080) protein levels and its associated signature. Functional studies validated increased Myc activity as a potent contributor to the impaired differentiation and enhanced renewal of neural stem cells doubly null for p53 and Pten (p53-/-Pten-/-) as well as tumor neurospheres derived from this model. Myc also serves to maintain robust tumorigenic potential of p53-/-Pten-/- tumor neurospheres. These murine modeling studies, together with confirmatory transcriptomic/promoter studies in human primary glioblastoma, validated a pathogenetic role of a common tumor suppressor mutation profile in human primary glioblastoma and established Myc as an important target for cooperative actions of p53 and Pten in the regulation of normal and malignant stem/progenitor cell differentiation, self-renewal, and tumorigenic potential.
* Structure Information
1. Primary Information
Length: 341 aa
Average Mass: 37.826 kDa
Monoisotopic Mass: 37.801 kDa
2. Domain Information
Annotated Domains: interpro / pfam / smart / prosite
Computationally Assigned Domains (Pfam+HMMER):
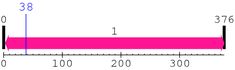
| domain name | begin | end | score | e-value |
|---|---|---|---|---|
| P53_TAD 1. | 5 | 29 | 46.4 | 1.1e-10 |
| --- cleavage 25 (inside P53_TAD 5..29) --- | ||||
| P53 1. | 95 | 289 | 511.1 | 1.4e-150 |
| P53_tetramer 1. | 318 | 339 | -13.6 | 9.4 |
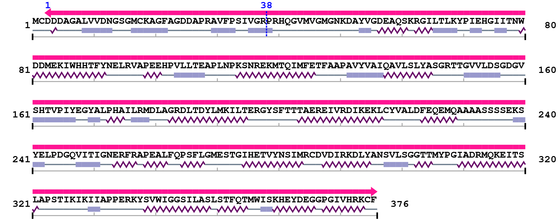
3. Sequence Information
Fasta Sequence: XSB2941.fasta
Amino Acid Sequence and Secondary Structures (PsiPred):
4. 3D Information
Not Available.
* Cleavage Information
1 [sites]
Cleavage sites (±10aa)
[Site 1] QETFSDLWKL25-LPENNVLSPL
Leu25  Leu
Leu
 |
|||||||||
| P10 | P9 | P8 | P7 | P6 | P5 | P4 | P3 | P2 | P1 |
|---|---|---|---|---|---|---|---|---|---|
| Gln16 | Glu17 | Thr18 | Phe19 | Ser20 | Asp21 | Leu22 | Trp23 | Lys24 | Leu25 |
 |
|||||||||
| P1' | P2' | P3' | P4' | P5' | P6' | P7' | P8' | P9' | P10' |
| Leu26 | Pro27 | Glu28 | Asn29 | Asn30 | Val31 | Leu32 | Ser33 | Pro34 | Leu35 |
 Sequence conservation (by blast)
Sequence conservation (by blast) Sequence conservation (by blast)
Sequence conservation (by blast)
| Reference peptide (cleaved bond±30 residues) |
|---|
| MEEPQSDPSVEPPLSQETFSDLWKLLPENNVLSPLPSQAMDDLMLSPDDIEQWFT |
Summary
| # | organism | max score | hits | top seq |
|---|---|---|---|---|
| 1 | Homo sapiens | 117.00 | 44 | p53 beta isoform |
| 2 | N/A | 117.00 | 14 | - |
| 3 | Pan troglodytes | 117.00 | 5 | PREDICTED: tumor protein p53 isoform 3 |
| 4 | synthetic construct | 117.00 | 3 | tumor protein p53 (Li-Fraumeni syndrome) |
| 5 | Tupaia belangeri chinensis | 117.00 | 1 | P53_TUPGB Cellular tumor antigen p53 (Tumor suppre |
| 6 | Macaca mulatta | 110.00 | 2 | tumor protein p53 |
| 7 | Chlorocebus aethiops | 110.00 | 1 | P53_CERAE Cellular tumor antigen p53 (Tumor suppre |
| 8 | Macaca fascicularis | 108.00 | 1 | P53 |
| 9 | Marmota monax | 89.40 | 1 | P53_MARMO Cellular tumor antigen p53 (Tumor suppre |
| 10 | Spalax judaei | 83.60 | 1 | p53 protein |
| 11 | Cavia porcellus | 82.40 | 1 | P53_CAVPO Cellular tumor antigen p53 (Tumor suppre |
| 12 | Delphinapterus leucas | 80.50 | 1 | P53_DELLE Cellular tumor antigen p53 (Tumor suppre |
| 13 | Meriones unguiculatus | 72.40 | 1 | p53 |
| 14 | Oryctolagus cuniculus | 71.20 | 1 | tumor protein p53 |
| 15 | Cricetulus griseus | 69.70 | 2 | P53_CRIGR Cellular tumor antigen p53 (Tumor suppre |
| 16 | Mesocricetus auratus | 69.70 | 1 | tumor supressor p53 |
| 17 | Sus scrofa | 68.20 | 3 | tumor suppressor p53 |
| 18 | Ovis aries | 67.80 | 1 | P53 protein |
| 19 | Canis familiaris | 67.80 | 1 | P53 |
| 20 | Felis catus | 66.20 | 2 | tumor protein p53 |
| 21 | Canis lupus familiaris | 65.90 | 1 | tumor protein p53 |
| 22 | Bos taurus | 64.30 | 2 | p53 |
| 23 | Mus musculus | 61.60 | 12 | transformation related protein 53 |
| 24 | Rattus norvegicus | 52.00 | 5 | tumor protein p53 |
| 25 | Bos primigenius | 50.40 | 1 | p53 gene product |
| 26 | Mastomys natalensis | 47.00 | 1 | p53 |
| 27 | Xenopus tropicalis | 44.70 | 1 | tumor protein p53 (Li-Fraumeni syndrome) |
| 28 | Xenopus laevis | 43.50 | 5 | p53 |
Top-ranked sequences
| organism | matching |
|---|---|
| Homo sapiens | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |||||||||||||||||||||||||#|||||||||||||||||||||||||||||| Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |
| N/A | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |||||||||||||||||||||||||#|||||||||||||||||||||||||||||| Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |
| Pan troglodytes | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |||||||||||||||||||||||||#|||||||||||||||||||||||||||||| Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |
| synthetic construct | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |||||||||||||||||||||||||#|||||||||||||||||||||||||||||| Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |
| Tupaia belangeri chinensis | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |||||||||||||||||||||||||#|||||||||||||||||||||||||||||| Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |
| Macaca mulatta | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |||||||||+|||||||||||||||#||||||||||||||+|||||||||+ || | Sbjct 1 MEEPQSDPSIEPPLSQETFSDLWKL#LPENNVLSPLPSQAVDDLMLSPDDLAQWLT 55 |
| Chlorocebus aethiops | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |||||||||+|||||||||||||||#||||||||||||||+|||||||||+ || | Sbjct 1 MEEPQSDPSIEPPLSQETFSDLWKL#LPENNVLSPLPSQAVDDLMLSPDDLAQWLT 55 |
| Macaca fascicularis | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWFT 55 |||||||||+|||||||||||||||#||||+|||||||||+|||||||||+ || | Sbjct 1 MEEPQSDPSIEPPLSQETFSDLWKL#LPENHVLSPLPSQAVDDLMLSPDDLAQWLT 55 |
| Marmota monax | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWF 54 ||| ||| |+||||||||||||| |#|||||||||+ | ||||+|| +|+| || Sbjct 1 MEEAQSDLSIEPPLSQETFSDLWNL#LPENNVLSPVLSPPMDDLLLSSEDVENWF 54 |
| Spalax judaei | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLS-PLPSQAMDDLMLSPDDIEQW 53 ||| ||| |+|||||||||||||||#||+||||| || +|+||+|||+|+ | Sbjct 1 MEEQQSDLSIEPPLSQETFSDLWKL#LPQNNVLSTPLSPNSMEDLLLSPEDVANW 54 |
| Cavia porcellus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 |||| || |+|||||||||||||||#|||||||| | || |+|||+++ | Sbjct 1 MEEPHSDLSIEPPLSQETFSDLWKL#LPENNVLSDSLSPPMDHLLLSPEEVASW 53 |
| Delphinapterus leucas | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 ||| |++ ||||||||||||||||#|||||+|| | |+|||+|||+|+ | Sbjct 1 MEESQAELGVEPPLSQETFSDLWKL#LPENNLLSSELSPAVDDLLLSPEDVANW 53 |
| Meriones unguiculatus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 ||||||| |+|||||||||||||||#|| |+|| | + |+||+| | |+ | Sbjct 1 MEEPQSDLSIEPPLSQETFSDLWKL#LPPKNLLSAL--EPMEDLLL-PQDVTSW 50 |
| Oryctolagus cuniculus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 ||| ||| |+|||||||||||||||#|||||+|+ + +||| || +|+ | Sbjct 1 MEESQSDLSLEPPLSQETFSDLWKL#LPENNLLTTSLNPPVDDL-LSAEDVANW 52 |
| Cricetulus griseus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLP-SQAMDDLMLSPDDIEQW 53 ||||||| |+| |||||||||||||#|| ||||| || | ++++| || +++ | Sbjct 1 MEEPQSDLSIELPLSQETFSDLWKL#LPPNNVLSTLPSSDSIEELFLS-ENVTGW 53 |
| Mesocricetus auratus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLP-SQAMDDLMLSPDDIEQW 53 ||||||| |+| |||||||||||||#|| ||||| || | ++++| || +++ | Sbjct 1 MEEPQSDLSIELPLSQETFSDLWKL#LPPNNVLSTLPSSDSIEELFLS-ENVAGW 53 |
| Sus scrofa | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVL-SPLPSQAMDDLMLSP 47 ||| ||+ ||||||||||||||||#|||||+| | | |++||+||| Sbjct 1 MEESQSELGVEPPLSQETFSDLWKL#LPENNLLSSELSLAAVNDLLLSP 48 |
| Ovis aries | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 ||| |++ |||||||||||||| |#|||||+|| | +|||+ +|+ | Sbjct 1 MEESQAELGVEPPLSQETFSDLWNL#LPENNLLSSELSAPVDDLLPYSEDVVTW 53 |
| Canis familiaris | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 |+||||+ +++|||||||||+|| |#|||||||| |+|+|+| |+ + | Sbjct 1 MQEPQSELNIDPPLSQETFSELWNL#LPENNVLSSELCPAVDELLL-PESVVNW 52 |
| Felis catus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 |+|| + ++||||||||||+|| |#|||||||| | ||++| || +|+ | Sbjct 1 MQEPPLELTIEPPLSQETFSELWNL#LPENNVLSSELSSAMNELPLS-EDVANW 52 |
| Canis lupus familiaris | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 ||| ||+ +++|||||||||+|| |#|||||||| |+|+|+| |+ + | Sbjct 1 MEESQSELNIDPPLSQETFSELWNL#LPENNVLSSELCPAVDELLL-PESVVNW 52 |
| Bos taurus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 ||| |++ +|||||||||||||| |#|||||+|| | +||| | |+ | Sbjct 1 MEESQAELNVEPPLSQETFSDLWNL#LPENNLLSSELSAPVDDL-LPYTDVATW 52 |
| Mus musculus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQWF 54 ||| ||| |+| |||||||| ||||#|| ++|| ||||+| | |+|++| Sbjct 4 MEESQSDISLELPLSQETFSGLWKL#LPPEDILS--SPHCMDDLLL-PQDVEEFF 54 |
| Rattus norvegicus | Query 1 MEEPQSDPSVEPPLSQETFSDLWKL#LPENNVLSPLPS---QAMDDLMLSPDDIE 51 ||+ ||| |+| |||||||| ||||#|| +++| + +|+|| | | | Sbjct 1 MEDSQSDMSIELPLSQETFSCLWKL#LPPDDILPTTATGSPNSMEDLFLPQDVAE 54 |
| Bos primigenius | Query 13 PLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPDDIEQW 53 ||||||||||| |#|||||+|| | +||| | |+ | Sbjct 1 PLSQETFSDLWNL#LPENNLLSSELSAPVDDL-LPYTDVATW 40 |
| Mastomys natalensis | Query 13 PLSQETFSDLWKL#LPENNVLSPLPSQAMDDLMLSPD 48 ||||||| ||||#|| ||| +||++ |||| Sbjct 2 PLSQETFQRLWKL#LPPEAVLSEASPNSMDNMFLSPD 37 |
| Xenopus tropicalis | Query 3 EPQSDPSVEPPLSQETFSDLWKL#LPE 28 || |+ +||||||||| ||| |#||+ Sbjct 2 EPSSETGMEPPLSQETFEDLWSL#LPD 27 |
| Xenopus laevis | Query 3 EPQSDPSVEPPLSQETFSDLWKL#LPE 28 || |+ ++|||||||| ||| |#||+ Sbjct 2 EPSSETGMDPPLSQETFEDLWSL#LPD 27 |
* References
[PubMed ID: 19434453] Chien WP, Wong RH, Wu TC, Cheng YW, Chen CY, Lee H, Potential increase in the prognostic value of p53 mutation by Pro72 allele in stage I non-small-cell lung cancer. Ann Surg Oncol. 2009 Jul;16(7):1918-24. Epub 2009 May 12.
[PubMed ID: 19423538] ... Koshiol J, Hildesheim A, Gonzalez P, Bratti MC, Porras C, Schiffman M, Herrero R, Rodriguez AC, Wacholder S, Yeager M, Chanock SJ, Burk RD, Wang SS, Common genetic variation in TP53 and risk of human papillomavirus persistence and progression to CIN3/cancer revisited. Cancer Epidemiol Biomarkers Prev. 2009 May;18(5):1631-7.
[PubMed ID: 19430562] ... Cho YH, Kim DY, Kim JH, Kim YM, Kim KR, Nam JH, Kim YT, Mutational analysis of KRAS, BRAF, and TP53 genes of ovarian serous carcinomas in Korean women. Yonsei Med J. 2009 Apr 30;50(2):266-72.
[PubMed ID: 19432615] ... Firouzabadi RD, Ghasemi N, Rozbahani MA, Tabibnejad N, Association of p53 polymorphism with ICSI/IVF failure and recurrent pregnancy loss. Aust N Z J Obstet Gynaecol. 2009 Apr;49(2):216-9.
[PubMed ID: 19426493] ... Galli P, Cadoni G, Volante M, De Feo E, Amore R, Giorgio A, Arzani D, Paludetti G, Ricciardi G, Boccia S, A case-control study on the combined effects of p53 and p73 polymorphisms on head and neck cancer risk in an Italian population. BMC Cancer. 2009 May 8;9:137.
[PubMed ID: 1394225] ... Somers KD, Merrick MA, Lopez ME, Incognito LS, Schechter GL, Casey G, Frequent p53 mutations in head and neck cancer. Cancer Res. 1992 Nov 1;52(21):5997-6000.
[PubMed ID: 1327751] ... Crook T, Vousden KH, Properties of p53 mutations detected in primary and secondary cervical cancers suggest mechanisms of metastasis and involvement of environmental carcinogens. EMBO J. 1992 Nov;11(11):3935-40.
[PubMed ID: 1303181] ... Bhatia K, Gutierrez MI, Magrath IT, A novel mutation in the p53 gene in a Burkitt's lymphoma cell line. Hum Mol Genet. 1992 Jun;1(3):207-8.
[PubMed ID: 1349102] ... Crook T, Wrede D, Tidy JA, Mason WP, Evans DJ, Vousden KH, Clonal p53 mutation in primary cervical cancer: association with human-papillomavirus-negative tumours. Lancet. 1992 May 2;339(8801):1070-3.
[PubMed ID: 1349175] ... Iavarone A, Matthay KK, Steinkirchner TM, Israel MA, Germ-line and somatic p53 gene mutations in multifocal osteogenic sarcoma. Proc Natl Acad Sci U S A. 1992 May 1;89(9):4207-9.